生物信息数据结构
FASTA格式
因为要便于存储生物序列信息,比如ACGT组成的DNA序列、蛋白质序列,生物信息学家们便基于 txt 文本格式定义了有一定规范的FASTA和FASTQ格式。
定义: - FASTA格式又称Pearson格式,是一种最简单的序列文件格式。最初由Pearson与Lipman一起于1988年首次提出,用于序列数据快速处理和存储。 - FASTA最后一个字母A表示联配(alignment)。
格式: -
第一行为>开头的标题行,包含序列的名称或唯一标识符信息 -
标题行和注释后,则是一行或多行构成的蛋白质或核酸序列数据,每行的字符串长度短于80个字符
1
2
3>gi|31563518|ref|NP_852610.1| microtubule-associated proteins 1A/1B light chain 3A isoform b [Homo sapiens]
MKMRFFSSPCGKAAVDPADRCKEVQQIRDQHPSKIPVIIERYKGEKQLPVLDKTKFLVPDHVNMSELVKI
IRRRLQLNPTQAFFLLVNQHSMVSVSTPIADIYEQEKDEDGFLYMVYASQETFGFIRENE
FASTQ格式
定义:一种用于存储生物序列(通常是核苷酸序列)及相应测序信息质量得分的文本文件。FASTQ格式与FASTA格式类似,但比FASTA格式多了序列的质量信息,序列字母和质量得分都用单个ASCII字符表示。
1. FASTQ文件命名规则:
Illumina测序仪下机FASTQ命名为(NextSeq CN500下机数据为bcl格式,经过bcl2fastq转化后名称类似),例如: Samplexx_S53_L002_R1_001.fastq.gz
Samplexx:样本名,与上机时在sampleSheet中填写的一致; S53:S后跟的数字与样本在sampleSheet中的顺序一致,从1开始; L002:L00,lane编号; R1:R,R1表示read1,R2表示read2。R1和R2为paired end reads。同一个样本的配对的FASTQ,只有这个地方不同; 001:001,通常为001;
Undetermined_S0_L001_R1_001.fastq.gz:存储index不匹配的reads
2. FASTQ文件格式
总共四列如下: 1
2
3
4@SIM:1:FCX:1:15:6329:1045:GATTACT+GTCTTAAC 1:N:0:ATCCGA
TCGCACTCAACGCCCTGCATATGACAAGACAGAATC
+
<>;##=><9=AAAAAAAAAA9#:<#<;<<<????#=
- 第一行,Sequence identifier
序列标识以及相关的描述信息,例如: @SIM:1:FCX:1:15:6329:1045:GATTACT+GTCTTAAC 1:N:0:ATCCGA
官网给的格式解释如下: 
1 | |
- 第二行,Sequence
- 序列信息,例如TCGCACTCAACGCCCTGCATATGACAAGACAGAATC
- 第三行,Quality score identifier line (consisting
only of a +)
- 以“+”开头,为节省存储空间什么也不加,以前会重复第一行信息
- 第四行,Quality score 测序质量值
- 长度与序列信息相同,且一一对应。
3. FASTQ质量值计算
Fastq格式中第四行的质量得分与碱基错误率的关系 (1)ASCII与碱基质量的关系
\[ ASCII=序列质量+33 \]
(2)碱基质量\(Q_{phred}\)和碱基错误率\(e\)的关系
\[ Q_{phred}=-10lge \]
- 关系:ASCII值越大,序列质量越好,碱基错误率越低
- 其中图片中最左侧的符号
!的ASCII值为33,而最右侧的符号~的ASCII为126。这也就确定了!对应的碱基错误率为\(100\%\),而~对应的碱基错误率为\(10^{-9.3}\)。(已经是极小的碱基错误率,相当于完全正确) - 规律:需要记住的大致比较ASCII:数字<大写字母<小写字母(小写字母ASCII = 大写字母ASCII + 32)
- 质量体系值phred的标准,在Solexa,Illumina(版本1.8及以上)采用Phred33;早期版本(<1.3)采用phred64


BED格式
BED (Browser Extensible Data)是一种灵活的储存数据的格式,主要用来储存基因组特征或注释信息。包括3列必选信息(chrom、chromStart 和 ChromEnd)和9列备选信息。BED 文件可用于 UCSC 的 Genome Browser 可视化工具。
BED格式有3列信息必选:
- 1. chrom - 染色体名称 (e.g. chr3, chrY, chr2_random) 或 scaffold (e.g. scaffold10671).
- 2. chromStart - 染色体或scaffold起始位点. 注意!是从0开始计数!
- 3. chromEnd - 染色体或scaffold终止位点. 注意!结束位点的碱基不包含在内。比如 chromStart=0, chromEnd=100, 那么实际上表示为0-99.
还有9列备选信息 - 4. name - BED的名称. - 5. score - 分数为0到1000。颜色越深,分数越高。 - 6. strand - 链方向. "." (=no strand) 或"+" 或 "-". - 7. thickStart - 绘制特征的起始位置(例如起始密码子)。当没有thick部分时,thickStart和thickEnd通常设置为chromStart位置。 - 8. thickEnd - 绘制特征的终止位置(例如终止密码子)。 - 9. itemRgb - RGB值 (e.g. 255,0,0). 如果track 行 itemRgb 参数设置为 "On", RBG值会决定BED文件中该行数据的颜色. - 10. blockCount - blocks (exons)在BED line的数量. - 11. blockSizes - 用逗号分隔的列表,用来表示block的的大小。列表中数字的数目应和_blockCount_ 一致。 - 12. blockStarts - 用逗号分隔的列表,用来表示block的起始。 所有_blockStart_ 的位点应该和_chromStart_ 相关。 列表中数字的数目应和_blockCount_ 一致。
BEDn文件的后缀n表示有几列信息,最少为BED3,最长为BED12
对于BED文件的处理,可以使用bedtools对储存了基因组特征信息的BED文件进行比较、注释、转化等操作。
GFF格式
定义: GFF(General Feature Format)格式是 Sanger 研究所定义的一种简单方便的数据格式,对 DNA 、RNA 及蛋白质序列的特征进行描述。GFF 格式由纯文本的9列信息按照制表符进行分割。其在 GMOD 中经常用于数据交换和基因组数据的表示。 General Feature Format (GFF)格式,和BED格式一样,也可用于储存基因组或蛋白质特征,主要用来注释基因组。
![樊龙江. 生物信息学(第2版).[M].浙江.科学出版社.2021:447.](https://bacon-bioinfomatic.oss-cn-beijing.aliyuncs.com/%E6%8B%BC%E5%9B%BE_20230720_203946_1%20442.jpg)
GTF格式
Gene Transfer Format (GTF),是一种基因组学中用于标记基因在基因组中位置并同时提供一些基本注释信息的文件,和GFF类似,都有9列数据并用制表符分隔。 Ensembl、GENCODE,以及UCSC基因组浏览器等项目都提供常见模式生物基因组的GTF注释文件。 但GTF格式主要是用来对基因进行注释,目前广泛使用的是第二版GTF2。
GFF和GTF格式的区别:
1.第3列feature/type不同;GFF可以为任意名称,而GTF则必须注明 2.第9列attribures不同;GFF键和值是以等号=隔开,GTF则是以空格隔开 参考:[生信资料 3] 生物信息学常见数据格式,汇总! - 知乎 (zhihu.com)
GFF第3列是type常见类型有:gene、exon、intron等;GTF第3列是feature,常见类型有:gene、transcript、CDS、5'UTR、start_codon、exon、stop_codon、3'UTR。 GTF第9列的attributes必须有gene_id和transcript_id这两个属性,GFF则不需要。
可以使用cufflinks中的gffread转换GFF和GTF:
1 | |
GBFF格式,GeneBank格式
GBFF格式为GeneBank数据库使用的记录格式。GBFF格式整体分为三部分,分别为描述部分、注释部分和序列部分。
![樊龙江. 生物信息学(第2版).[M].浙江.科学出版社.2021:447.](https://bacon-bioinfomatic.oss-cn-beijing.aliyuncs.com/%E6%8F%8F%E8%BF%B0%E9%83%A8%E5%88%86%E5%8C%85%E6%8B%AC%E4%BA%86%E6%95%B4%E4%B8%AA%E8%AE%B0%E5%BD%95%E7%9A%84%E7%9B%B8%E5%85%B3%E4%BF%A1%E6%81%AF.jpg)
问题1:辨析描述部分中Locus和Accession、Version、GI - Accession是在数据库中唯一确定且不变的,即使数据提交者改变了数据内容,也不改变。 - Locus是一条特定序列的名字,不一定和 Accession 一致(如果研究人员在提交序列时没有给序列命名,则检索号和 Locus 是一样的)。 - Locus 相当于是序列的名字,而 Accession 是序列的学号,在不同的数据库中,Locus 一致,但 Accession 不一样。 - Version是序列更新时,在检索号的后面加一个“.数字”,表示第几次修改 - GI 号,在 version 号的后面,且序列更新就会被赋予一个新的 GI 号。 综上所述:Locus和Accession唯一且不变,但Locus和Accession不一定相同;Version和GI不唯一且随序列更新而改变。
问题2:浏览真核生物的mRNA序列,如何知道是什么类型的mRNA、编码的蛋白质产物、mRNA序列中信号肽的位置和真正编码蛋白质的位置 - 通过CDS中的note可以知道mRNA的类型 - 通过sig_peptide可以知道mRNA中编码信号肽的位置 - 通过mat_peptide可以知道编码成熟蛋白质的位置,其终止位置会比CDS的终止位置少三个碱基(终止密码子) - 有些时候还有misc_feature表示该位置的序列是推测的,并不是由实验证实的。
问题3:浏览真核生物的DNA序列,怎么知道被分为了几个片段、在染色体上的位置、Gene拼接方式、转录的成熟mRNA序列、是第几个外显子 - 通过第一部分中SEGMENT可以知道基因被分为了几个片段,且该序列属于第几个片段 - 通过FEATURE中map可以知道序列在染色体上的位置 - 通过GENE可以知道拼出完整基因所需的所有片段的检索号以及检索号之间的拼接方式(从哪个检索号的哪一部分片段到下一个检索号的哪一部分片段到...) - 通过mRNA部分可以知道该基因经过剪切成熟后的mRNA序列,其可能含有不止一条序列,比如可能有含有信号肽mRNA和不含信号肽的mRNA - 通过exon部分可以知道该序列中外显子的位置以及属于这个基因的第几个外显子。
SRA格式
定义: - Sequence Read Archive (SRA) 数据库,由国家生物技术信息中心(NCBI)于2007年建立,旨在为RNA-Seq和ChIP-Seq研究以及包括人类微生物组计划和1000基因组计划在内的大规模研究产生的数据提供存储库。 - 用于储存来自不同高通量测序平台的原始序列数据和比对信息。SRA为DNA测序数据提供公共存储库,特别是高通量测序生成的“短读长”,其长度通常小于1,000bp。 - SRA是国际核苷酸序列数据库协作(INSDC)的一部分,由NCBI、欧洲生物信息学研究所(EBI)和日本DNA数据库(DDBJ)合作运行。 - 提交给SRA的文件的首选数据格式是BAM格式,它能够存储对齐和未对齐的读序。SRA内部依赖于NCBI SRA工具包,该工具包用于所有三个INSDC成员数据库,以提供灵活的数据压缩、API访问和转换为其他格式,如FASTQ。
SRA数据库用6种不同的前缀区分不同的数据: 1. SRA是提交数据时得到的数据号,包含下列5种前缀所组成的数据集。 2. SRP表示 SRA Studies/projects;研究课题或项目的元数据;一个Study(SRP)可能包含多个experiments。 3. SRX 表示 Experiments;包含元数据及其建库方式、测序平台、处理方式。 4. SRR 表示 Runs;包含真实的测序数据;一个Experiment(SRX)可能有多个Run。 5. SRS 表示 Samples;描述进行测序的物理样本的元数据。 6. SRZ 表示 Analysis;包含序列数据分析的BAM文件和描述序列分析的元数据。
SRA中数据结构的层次关系为:Studies->Experiments->Samples->Runs。这样分类是以便更好的找到所需信息。
SAM格式
定义: - SAM是一种用于保存映射到参考序列的短读序的比对信息同时也可能包含未映射序列信息的文件格式。 - 它通常以标题部分开头,然后是每个读序的制表符分隔行的比对信息。 - SAM文件经过转化为二进制BAM文件后,可用于下游生物信息学分析,如基因差异性表达分析,调查生物多样性,分析DNA甲基化或调查DNA-蛋白质相互作用。
SAM文件由header信息和mapping信息两部分组成 - 头部区:以'@'开始,体现了比对的一些总体信息。比如比对的SAM格式版本,参考基因组信息,比对的参考序列,比对使用的软件以及注释行等。 - 主体区:比对结果,每一个比对结果是一行,有11个主列和一个可选列。

@HD,说明符合标准的版本、对比序列的排列顺序; @SQ,参考序列说明; @RG,比对上的序列(read)说明; @PG,使用的程序说明; @CO,任意的说明信息。

对于mapping结果,主要看 - 第二列,如果是0则是正常的,如果是4则是没有map上,16表示map到的反转链上的 - MD表示未匹配的碱基信息,如MD:Z:3C30T3,表示从左往右前三个碱基mapped,第四个碱基在参考基因组上是C而在此序列中为A,然后再有30个碱基mapped上,倒数第四个碱基在参考基因组上是T而在此序列中为G - NM表示错配的数量。(可以先看NM来确定错配的数量,再看MD错配信息)
VCF格式
定义: Variant Call Format,VCF是一种标准化的文本文件格式,用于表示SNP、indel和结构变异调用。VCF规范曾经由1000基因组计划维护,但其管理和进一步开发已由全球基因组学与健康联盟的基因组数据工具包团队接管。
每列数据的说明: - 第一列 CHROM:染色体 - 第二列 POS:基因组位置 -
第三列 ID:变异位点的rsID号,如果没有的话用"."表示
- 第四列 REF:与参考基因组一样的位点
- 第五列 ALT:与参考基因组不一样的位点 - 第六列
QUAL:call出这个位点的质量。这个值等于-10log10(p),p值是call错alt
allele错误的概率。也就是QUAL越大出错概率越小
- 第七列
FILTER:对变异位点进行过滤,如果通过则为PASS,如果没有进行过滤就是"." -
第八列 INFO:这一列是额外信息 - 第九列
FORMAT:最后是比较让人注意的Genotype也就是基因型等的信息,比较重要的是GT,AD,DP,PL,GQ:
第八列: AC,AF 和 AN:AC(Allele Count) 表示该Allele的数目;AF(Allele Frequency) 表示Allele的频率; AN(Allele Number) 表示Allele的总数目。 \[ AF=\frac{AC}{AN} \] DP:reads覆盖度。是一些reads被过滤掉后的覆盖度。
Dels:Fraction of Reads Containing Spanning Deletions。进行SNP和INDEL calling的结果中,有该TAG并且值为0表示该位点为SNP,没有则为INDEL。
FS:使用Fisher’s精确检验来检测strand bias而得到的Fhred格式的p值。该值越小越好。一般进行filter的时候,可以设置 FS < 10~20。
第九列: GT,即genotype,表示为0/1, 1/1, 0/0或者是0|1, 1|0, 0|0, 1|2等。其实 - 0代表REF allele - 1代表第一个ALT allele - 2代表第二个ALT allele
比如REF是A,ALT是C,T(有两个ALT)。某个人是A/C,那么基因型就是0/1,A/T的话就是0/2,C/C就是1/1,以此类推。此外还可能见到0|1或者1|1中间是竖线不是斜线的情况,这种是已经phased的genotype,也就是已经知道REF/ALT allele是来自于父亲还是母亲了。比如有的数据库的phased的数据是|前的是父亲的allele,|后的是母亲的allele。比如REF是A,ALT是C,T;基因型为1|0,则父亲是第一个ALT也就是C,母亲是REF也就是A。不过对于有的phased数据而言第一个并不一定是父亲。 参考:DNA 5. 基因组变异文件VCF格式详解 - 知乎 (zhihu.com)
AD: REF和ALT allele的深度,以逗号作为分隔,前面表示覆盖到ref上的reads数,后面则是覆盖到ALT allele的reads数
DP:这个位点的深度。
GQ:表示最可能的基因型的质量值。表示的意义同QUAL。Phred格式(Phred_scaled)的质量值,表示在该位点该基因型存在的可能性;该值越高,则Genotype的可能性越大;计算方法:\(Phred值 = -10 * log (1-p)\) ,p为基因型存在的概率。
PL:对应3个以逗号隔开的值,这三个值分别表示该位点基因型是0/0,0/1,1/1的没经过先验的标准化Phred-scaled似然值(L)。这三种指定的基因型(0/0,0/1,1/1)的概率总和为1。如果转换成支持该基因型概率(P)的话,由于\(L=-10lgP\),那么\(P=10^{(-L/10)}\),因此,当L值为0时,\(P=10^0=1\)。因此,这个值越小,支持概率就越大,也就是说是这个基因型的可能性越大。
1 | |
在这个位点,GT=0/1,也就是说这个位点的基因型是C/T;GQ=25.92,质量值并不算太高,可能是因为cover到这个位点的reads数太少,DP=4,也就是说只有4条reads支持这个地方的变异;AD=1,3,也就是说支持REF的read有一条,支持ALT的有3条;在PL里,这个位点基因型的不确定性就表现的更突出了,0/1的PL值为0,虽然支持0/1的概率很高;但是1/1的PL值只有26,也就是说还有10^(-2.6)=0.25%的可能性是1/1;但几乎不可能是0/0,因为支持0/0的概率只有\(10^{-10.3}=5\times 10-11\)。 参考:生物基因数据文件——vcf格式详解_vcf官方说明_vickyleexy的博客-CSDN博客
总结: - AD前后相加=DP; - DP越大,覆盖深度越大; - GQ就一个值,GQ越大,基因型质量越高; - PL有三个值,每个位置的PL越小,该位置基因型的可能性越大(前,中,后分别为0/0,0/1,1/1)
PDB格式
定义: The Protein Data Bank(PDB)文件格式是描述蛋白质数据库中保存的分子三维结构的文本文件格式。PDB格式提供了蛋白质和核酸结构的描述和注释,包括原子坐标、二级结构分配以及原子连通性。此外,实验元数据也被存储。PDB格式是蛋白质数据库的传统文件格式,它现在以较新的mmCIF文件格式保存生物大分子的数据。
由6个部分组成 - 第一部分:头信息 - HEADER:蛋白质结构的基本信息描述,包括分子类别,存储日期,PDB ID - TITLE:结构的标题 - COMPND:对结构中各个分子的描述。从这里可以看出 3H6X 这个结构是由三条链形成 的三聚体结构。 - SOURCE:结构中所包括的每一个分子的实验来源。 - KEYWDS:用于数据库搜索的关键词 - EXPDTA:测定结构所采用的实验方法。PDB中绝大部分结构都是通过 X 射线衍射法测定的,少数是核磁共振法,极少数是使用包括电子显微镜在内的其他方法测定的。 - AUTHOR:作者信息 - REVDAT:历史上曾经对该数据库记录进行过的修改。 - JRNL:发表结构的文献信息。 - REMARK:无法归入其他部分的注释。 - 第二部分:一级结构信息 - DBREF:该蛋白质在蛋白质序列数据库里的检索号等信息。 - SEQRES:氨基酸序列。 - MODRES:对标准残基上的修饰,比如第 56 号位置的蛋氨酸被硒代蛋氨酸所取代。 - 第三部分:非标准残基信息 - HET:非标准残基及位置。 - HETNAM:非标准残基的化学名称。 - FORMUL:非标准残基的化学式。 - 第四部分:二级结构信息 - HELIX:位于螺旋结构上的氨基酸所在位置及所属链。 - SHEET:位于折片结构上的氨基酸所在位置及所属链。 - TURN:位于转角结构上的氨基酸所在位置及所属链。 - 第五部分:实验参数信息 - CRYST1:晶胞参数。 - ORIGXn:直角-PDB 坐标。 - SCALE*:直角部分结晶学坐标。 - 第六部分:3D坐标信息 - ATOM:每个原子的3D坐标信息 - CONECT:原子间化学键连接信息。 - MASTER:版权拥有者信息。 - END:结束符。


在人为浏览PDB文件的时候,重点关注其HEADER部分的分子类型;COMPND的有几条链;EXPDTA所描述的使用了哪种测定蛋白质结构的方法; PDB文件中最重要的便是原子3D坐标信息 参考:生物数据库之一级蛋白质数据库 - 简书 (jianshu.com)
UniprotKB
UniprotKB下载蛋白质序列
下面以 PO5F1_HUMAN 为例,下载对应的fasta序列来看看:
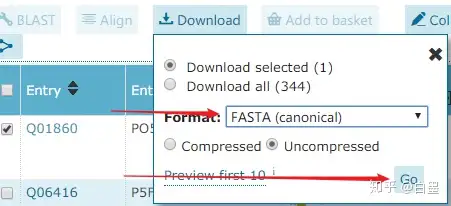
下载到的序列:
1 | |
首先看 > 后的注释信息
- sp:Swiss-Prot数据库的简称,也就是上面说的验证后的蛋白数据库
- Q01860:UniProt ID号
- PO5F1_HUMAN:是UniProt 的登录名
- POU domain, class 5, transcription factor 1:蛋白质名称
- OS=Homo sapiens:OS是Organism简称,Homo sapiens为人的拉丁文分类命名,也就是这是人的蛋白质
- OX=9606:Organism Taxonomy,也就是物种分类数据库Taxonomy ID
- GN=POU5F1:Gene name,基因名为POU5F1
- PE=1:Protein
Existence,蛋白质可靠性,对应5个数字,数字越小越可靠:
- 1:Experimental evidence at protein level,蛋白质水平的实验证据
- 2:Experimental evidence at tranlevel,转录水平的实验证据
- 3:Protein inferred from homology,同源推测出的蛋白
- 4:Protein predicted,预测的蛋白质
- 5:Protein uncertain,未知的蛋白质
- SV=1:Sequence Version,序列版本号
对于UniprotKB数据库进行检索,可以知道一个基因对应的蛋白质。 其中Entry表示蛋白质学历的在UniprotKB数据库中的检索号,Entry_Name是检索名,二者都是对于一条蛋白质序列的唯一标识(区分,UPnirotKB中检索号为Entry,GeneBank中是Accession) UniprotKB可以对序列进行比对、格式转换和存储 也可以查看蛋白质序列的注释信息
| 类型 | 描述 |
|---|---|
| Function | 蛋白质的功能,生化反应方程式、代谢途径 |
| Name&Taxomomy | 蛋白质的各种名字,所属物种和分类学谱系 |
| Subcellular location | 蛋白质的细胞定位,如果一个蛋白质存在两个异构体,则在亚细胞定位上就会存在两种定位类型 |
| Pathology&Biotechnology | 蛋白质突变或缺失导致的疾病及表型信息 |
| PTM/Processing | 蛋白质翻译后修饰或翻译后加工的信息 |
| Expression | 基因在mRNA水平的表达信息 |
| Interaction | 蛋白质之间的互作信息,包括互作蛋白的链接和在各种蛋白质互作网络数据库的记录链接 |
| Structure | 蛋白的二级和三级结构信息,注意🤔只有已通过实验验证并提交给PDB数据库的蛋白质才有结构注释 |
| Family&Domains | 蛋白质家族及结构域信息 |
| Sequence | 蛋白质氨基酸序列信息 |
| Cross-references | 通往其他含有该蛋白质的数据库链接 |
| Publications | 关于这个蛋白质已发表的文献信息 |
| Entry-information | 这条数据库记录的录入信息 |
| Miscellaneous | 剩余杂项信息 |
| Similar proteins | 在UniRef数据库里找到与该蛋白质序列水平相似的其他蛋白质 |